Time-Dependent Many-Body Theory and Quantum Electronic Dynamics
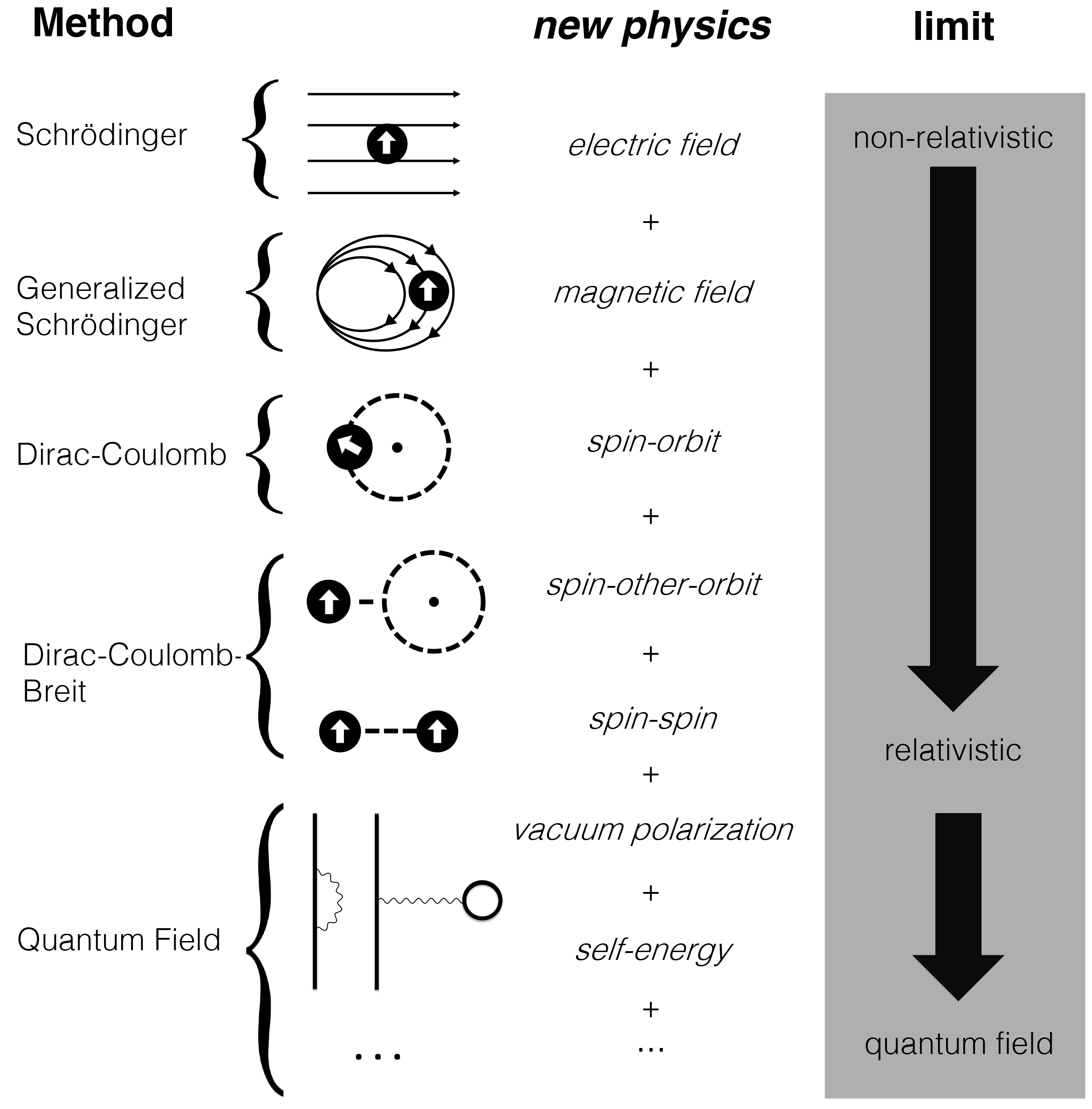
The time-dependent Schrödinger and Dirac equations govern all non-equilibrium quantum mechanical processes of a many-electron system. Understanding these processes is crucial to many advanced scientific research and technological developments. One area of the research effort in the Li group focuses on the development of time-dependent many-electron theories and computational methods that can be applied to simulate realistic and chemically relevant non-equilibrium many-electron phenomena driven by external perturbations (electromagnetic fields and dielectric media) and internal spin couplings (spin-spin, spin-orbit and spin-other-orbit). Several specific time-dependent many-body formalisms that are being developed in the Li research group include (1) time-dependent Hartree-Fock (TDHF) and time-dependent density functional theory (TDDFT), (2) time-dependent non-collinear generalized TDHF and TDDFT, and (3) time-dependent two- and four-component relativistic Dirac formalism. These new spin- and time-dependent many-electron methods will open many new avenues in simulating many-electron dynamics, such as ultrafast charge-transfer, spin transport, spin-crossover, and electronic (and spin) decoherence and dephasing etc.
Relativistic Electronic Structure Theory
For many molecules, such as those containing heavy elements, the non-relativistic Schrodinger equation is no longer valid. To go beyond the Schrodinger equation, we are developing methods based off variational treatments of the relativistic Dirac equation. This allows us to accurately treat relativistic interactions, such as spin-orbit coupling, for molecular systems. In particular, we are interested in variational two-component treatments of the Dirac equation, which allow us to extend the methods to excited state and non-equilibrium phenomena, which is important for predicting spectroscopic signatures of heavy elements and magnetic systems, as well as dynamic processes where the molecular spin state changes.
Low-Scaling Electronic Structure Theories and Optimization Algorithms
The size of systems being investigated using the tools of quantum chemistry is ever increasing. Discerning the equilibrium conformations of biomolecules, polymers and nanostructures with hundreds to thousands of atoms is often computationally demanding due to the nonlinear scaling of computational difficulty with respect to the number of electronic and nuclear degrees of freedom. Methods which improve upon the computational cost and general stability of conventional structure optimization and electronic structure algorithms would be widely appreciated.
In this area of research, we focus on developing fast, efficient methods for the accurate calculations of both ground and excited state molecular properties. We are also interested in modernizing these computational methods by utilizing the capabilities of advanced computer machinery, including high performance parallelism and cost effective sparse matrix manipulations. Several recently developed methods include a unique “density-of-states” Moller-Plesset theory (DOS-MPn), which accelerates electron correlation energy calculations on large conjugated polymers, a low-scaling equation-of-motion second order perturbation theory (EOM-MBPT2) which excels at describing Rydberg states, and and a highly parallelizable configuration interaction (CI) method, that extends the limit of high-accuracy first-principles calculations. We are also actively working on multi-reference methods in order to understand situations where standard single-reference black-box methods fail, such as near-degeneracies and conical intersections.
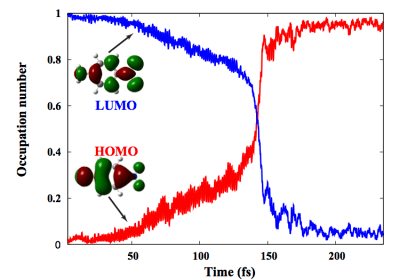
First-Principles Nonadiabatic Dynamics
Another active development area in the Li research group focuses on accurately reproducing chemical dynamics for a wide range of system sizes and time scales. To this end, we develop efficient numerical schemes to simultaneously integrate the quantum and classical equations of motion for all or some subset of the nuclear, electronic (spatial and spin), and bath degrees of freedom of chemical systems. Through the selective inclusion of interaction or coupling between these subsystems, the minimal-complexity models required to describe the physical phenomenon of interest are constructed. This modular approach allows maximum computational efficiency of a given investigation, since only the relevant physics are described by selectively treating the interaction between electron, nuclei, and bath at varying levels of accuracy (i.e. mean-field versus explicitly-interacting subsystems).
The influence of transitions between adiabatic electronic states on the propagation of the nuclear subsystem and vice versa can be included in dynamics in both state-specific and mean-field capacities through the surface hopping and Ehrenfest methodologies respectively. Furthermore, we aim to develop methods to treat spin dynamics at various levels of accuracy through integration of the time-dependent generalized Hartree-Fock/Kohn-Sham equations, with the eventual goal of a fully relativistic treatment of spin with a time-dependent Dirac-Fock formalism.
With these efficient methods in place, we can probe the dynamical properties of emerging spintronic and molecular plasmonic materials. Specific applications include the propagation and dephasing of non-equilibrium spin states in dilute magnetic semiconductors and plasmonic states induced in noble metal nanowires. Nonradiative relaxation pathways implicated in photochemical reactions are also open for investigation with tractable implementations of ab initio surface hopping methods.
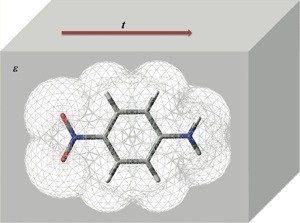
Real-Time Time-Dependent Dielectric Response
The time delay between photoexcitation of a solute and the electric polarization of the solvent is of enormous importance to the proper description of solvated excited state chemical dynamics. This out-of-phase dielectric response is responsible for the frequency dependence of dielectric media permittivities, and the dielectric relaxation/loss phenomenon. In conjunction with the real-time time dependent electronic structure theories that we have implemented, we are currently developing an extension of the PCM model in which the dielectric medium polarization carries explicit time dependence. In this case, the charge density at the interface of the embedded system and the dielectric medium becomes a dynamic variable, and solvent and solute time evolution are treated on an equal footing.
We have derived an equation of motion for the bath degrees of freedom based on first order time-dependent perturbation theory, which couples to (and is integrated simultaneously with) the equations of motion for the embedded system. With the lagging response of the solvent properly modeled, RT-TDDR represents a tractable method for the treatment of environmentally-induced decoherence, since the fluctuating dielectric polarization may either constructively or destructively interfere with certain components of the superposition state propagated in the embedded system. Furthermore, since polarization of the dielectric medium occurs at an energetic cost to the embedded system, energy can be freely exchanged with and/or dissipated to the bath, yielding accurate first-principle open-system dynamics for the embedded system.
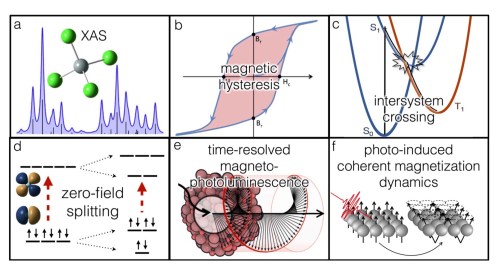
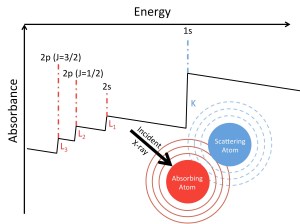
X-Ray Spectroscopy and High-Energy Chemical Physics
X-Ray absorption spectroscopy (XAS) and transient X-Ray absorption (XTA) are complex spectroscopies offering a wealth of information about electronic and nuclear structure. They also remain a challenging problem for theorists. Electronic relaxation due to the core excitations, environmental effects, scalar and spin-orbit relativistic effects, and higher-order multipole coupling to the incident field all need to be accounted for. An ongoing area of research in the Li group is the application of standard single-reference electronic structure methods to these problems through energy specific (ES) approaches. This allows these higher energy excitations to be probed without the need to solve for the lower energy excitations. Real-time TDDFT (RT-TDDFT) can also be applied to determine the effects of electronic relaxation due to the core excitation and non-equilibrium environmental effects.
Chemical Physics of Defects in Nanocrystals
Doping inorganic semiconductor quantum dots (QDs) with transition-metal ions introduces unique electronic, optical, and magnetic properties that are otherwise absent in the host semiconductor lattice. The potential to integrate room-temperature ferromagnetism with the electrical properties of semiconducting materials makes these so-called diluted magnetic semiconductors (DMSs) attractive for spin-electronic and spin-photonic applications. Our recent advances have revealed the microscopic origins of carrier-dopant and dopant-dopant exchange interactions and the effects of such interactions on the absorption spectra of these materials.
The overarching objective of this program is to computationally design and search for stable and controllable nano-sized DMS materials. Specifically, our research focuses on (i) the roles of defects, such as transition-metal and p-type dopants, in activating high Curie-temperature ferromagnetism; (ii) charge-transfer transitions for potential solar energy conversion; (iii) chemical and physical processes required to control magnetization using time-dependent quantum mechanical electrodynamics of charge-transfer delocalization, magnetic polaron formation, and magnetization reversal dynamics; and (iv) dopant-centered Auger-type processes and their role in the photodynamics of these doped nanocrystals.